Cryomicroscopy Enhances Mapping of Cystic Fibrosis Protein
By LabMedica International staff writers
Posted on 05 Apr 2017
A recent paper detailed the three-dimensional (3D) structure of cystic fibrosis transmembrane conductance regulator, the protein responsible for the genetic disorder cystic fibrosis (CF).Posted on 05 Apr 2017
The cystic fibrosis transmembrane conductance regulator (CFTR) is an ATP-binding cassette (ABC) transporter that uniquely functions as an ion channel. Investigators at The Rockefeller University used electron cryomicroscopy (cryo-EM) to determine the three-dimensional (3D) structure of dephosphorylated human CFTR at a resolution of 3.9 Angstroms.
.")
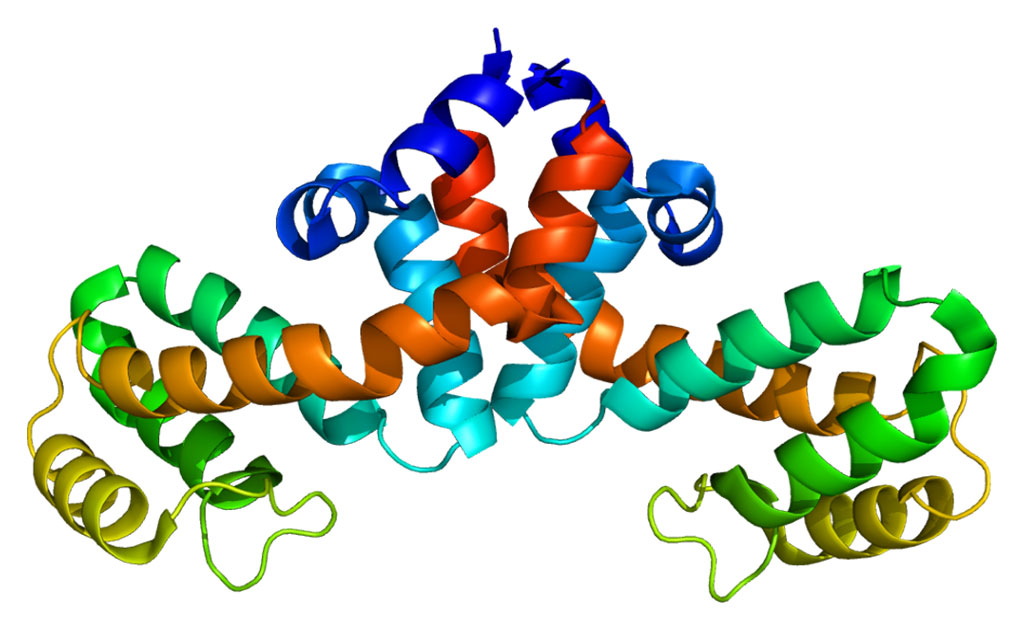
Image: The molecular structure of the human cystic fibrosis protein, which controls the flow of crucial ions in and out of cells (Photo courtesy of the Laboratory of Membrane Biology and Biophysics at The Rockefeller University).
Researchers have historically relied on NMR and X-ray diffraction techniques to determine the structures of molecular complexes and proteins that play a role in the causes of various disease states. Structural information about a variety of medically important proteins and drugs has been obtained by these methods. Cryo-EM is a complementary analytical technique that provides near-atomic resolution without requirements for crystallization or limits on molecular size and complexity imposed by the other techniques. Cryo-EM allows the observation of specimens that have not been stained or fixed in any way, showing them in their native environment while integrating multiple images to form a three-dimensional (3D) model of the sample.
Results published in the March 23, 2017, issue of the journal Cell revealed that the structure of human CFTR was quite similar to that determined previously for the zebrafish. This close resemblance reinforced its relevance for understanding CFTR function. The human CFTR structure generated by this study revealed a previously unresolved helix belonging to the R domain docked inside the intracellular vestibule, precluding channel opening. In addition, the three-dimensional (3D) map showed a feature distinguishing CFTR from all other ABC transporters: the helix-loop transition in transmembrane helix eight, which likely forms the structural basis for CFTR’s channel function.
"With these detailed new reconstructions, we can begin to understand how this protein functions normally, and how errors within it cause cystic fibrosis," said senior author Dr. Jue Chen, professor of membrane biology and biophysics at The Rockefeller University. "We now know that the conclusions we drew from our previous work in zebrafish also apply to us."
Platinum Member
COVID-19 Rapid Test
OSOM COVID-19 Antigen Rapid Test


POCT Fluorescent Immunoassay Analyzer
FIA Go

Gold Member
ADAMTS-13 Protease Activity Test
ATS-13 Activity Assay