Loss of Neuronal Enzyme Underlies Rare Childhood Epileptic Encephalopathy
By Gerald M. Slutzky, PhD
Posted on 08 Dec 2016
A recessive mutation causing loss of an enzyme required for neuronal development was shown to underlie a rare form of childhood epileptic encephalopathy.Posted on 08 Dec 2016
Epileptic encephalopathy, which is often linked to improper development of the brain, is a rare but devastating sub-form of epilepsy that results in severe mental and physical disabilities in children from birth. To better understand the genetic and mechanistic causes of this disease, investigators at McGill University (Montreal, Canada) and collaborators in Canada, Saudi Arabia, Jordan, and Germany performed whole exome sequencing on three children with epileptic encephalopathy from two families, one from Saudi Arabia and another from Jordan.
.")
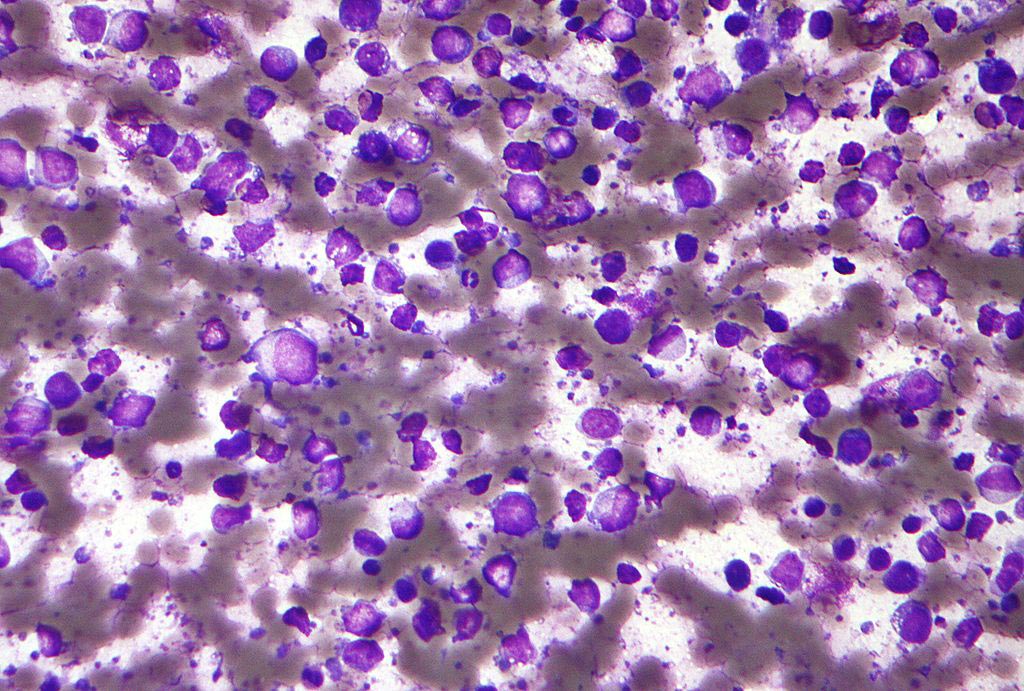
Image: A neuron in culture was transduced with a virus that expresses a green fluorescent protein and an inhibitory RNA that causes loss of the DENND5A protein. The neurons where then stained with a marker of neuronal processes in red (Photo courtesy of Peter McPherson Laboratory, Montreal Neurological Institute, McGill University).
The investigators reported in the November 17, 2016, online edition of the American Journal of Human Genetics that epileptic encephalopathy, which featured cerebral calcifications and coarse facial features, was caused by recessive loss-of-function mutations in the gene DENND5A.
DENND5A contains a DENN domain, an evolutionarily ancient enzymatic module conferring guanine nucleotide exchange factor (GEF) activity to multiple proteins serving as GEFs for Rab proteins, which are key regulators of membrane trafficking. DENND5A is detected predominantly in neuronal tissues, and its highest levels occur during development. Knockdown of DENND5A leads to striking alterations in neuronal development.
Mechanistically, changes caused by lack of DENND5A activity appeared to result from upregulation of neurotrophin receptors, leading to enhanced downstream signaling. Neurotrophins are a family of proteins involved in the survival, development, and function of neurons.
"Our study demonstrates the importance of membrane trafficking in neuronal development and it provides a new pathophysiological mechanism for this disease type. This will allow physicians around the world to test if mutations in DENND5A are causing the disease in their patients, and also to provide genetic counseling for affected families," said first author Dr. Chanshuai Han, a neurodegenerative disease researcher at McGill Univesity.
Related Links:
McGill University
Platinum Member
COVID-19 Rapid Test
OSOM COVID-19 Antigen Rapid Test


POCT Fluorescent Immunoassay Analyzer
FIA Go
Gold Member
Real-time PCR System
GentierX3 Series