Gene Editing Corrects Hemoglobin Defects in Beta-Thalassemia and Sickle Cell Disease
By LabMedica International staff writers
Posted on 25 Aug 2016
A team of hematology researchers used the CRISPR/Cas9 gene editing technique to correct the mutations that cause defective blood cell morphology in beta-thalassemia and sickle cell disease.Posted on 25 Aug 2016
CRISPRs (clustered regularly interspaced short palindromic repeats) are segments of prokaryotic DNA containing short repetitions of base sequences. Each repetition is followed by short segments of "spacer DNA" from previous exposures to a bacterial virus or plasmid. CRISPRs are found in approximately 40% of sequenced bacteria genomes and 90% of sequenced archaea. CRISPRs are often associated with cas genes that code for proteins related to CRISPRs. Since 2013, the CRISPR/Cas system has been used in research for gene editing (adding, disrupting, or changing the sequence of specific genes) and gene regulation. By delivering the Cas9 enzyme and appropriate guide RNAs into a cell, the organism's genome can be cut at any desired location. The conventional CRISPR/Cas9 system is composed of two parts: the Cas9 enzyme, which cleaves the DNA molecule and specific RNA guides (CRISPRs) that shepherd the Cas9 protein to the target gene on a DNA strand.
.")
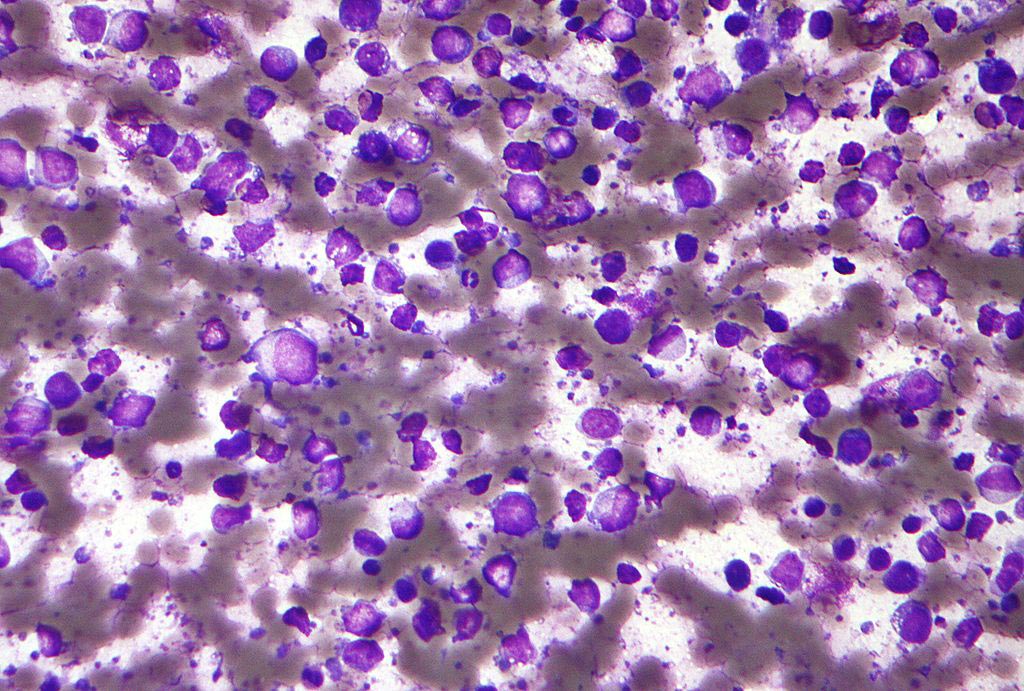
Image: Healthy blood cells along with sickle-cell diseased cells (Photo courtesy of Science Picture Co./Corbis).
Disorders resulting from mutations in the hemoglobin subunit beta gene (which encodes beta-globin), mainly sickle cell disease (SCD) and beta-thalassemia, become symptomatic after birth as fetal gamma-globin expression from two genes, hemoglobin subunit gamma 1 (HBG1) and HBG2, decreases and adult beta-globin expression increases. This shifts red blood cell (RBC) hemoglobin from the fetal to the adult form. These disorders are alleviated when postnatal expression of fetal gamma-globin is maintained, as the case of a benign genetic condition that causes high-level expression of fetal hemoglobin throughout life.
Investigators at St. Jude Children's Research Hospital (Memphis, TN, USA) reported in the August 15, 2016, online edition of the journal Nature Medicine that they had performed CRISPR–Cas9-mediated genome editing of human blood progenitors to mutate a 13-nucleotide sequence that was present in the promoters of the HBG1 and HBG2 genes, thereby recapitulating the naturally occurring "fetal hemoglobin for life" mutation. Edited progenitors produced RBCs with increased fetal hemoglobin levels that were sufficient to inhibit the pathological hypoxia-induced RBC morphology found in sickle cell disease.
"Our approach to gene editing is informed by the known benefits of hereditary persistence of fetal hemoglobin," said senior author Dr. Mitchell J. Weiss, professor of hematology at St. Jude Children's Research Hospital. "It has been known for some time that individuals with genetic mutations that persistently elevate fetal hemoglobin are resistant to the symptoms of sickle cell disease and beta-thalassemia, genetic forms of severe anemia that are common in many regions of the world. We have found a way to use CRISPR gene editing to produce similar benefits."
"Our work has identified a potential DNA target for genome editing-mediated therapy and offers proof-of-principle for a possible approach to treat sickle cell and beta-thalassemia," said Dr. Weiss. "We have been able to snip that DNA target using CRISPR, remove a short segment in a "control section" of DNA that stimulates gamma-to-beta switching, and join the ends back up to produce sustained elevation of fetal hemoglobin levels in adult red blood cells. Using genome editing to restore the hereditary persistence of fetal hemoglobin is an attractive possibility, because it can be achieved relatively easily using current technologies. The condition is known to be benign in people who inherit similar naturally occurring mutations."
Related Links:
St. Jude Children's Research Hospital
Platinum Member
COVID-19 Rapid Test
OSOM COVID-19 Antigen Rapid Test


Complement 3 (C3) Test
GPP-100 C3 Kit

Gold Member
Fully Automated Cell Density/Viability Analyzer
BioProfile FAST CDV